PBPK 模型的组成部分
-
PBPK 模型结构和假设
PBPK 模型由对应于体内不同组织的隔室组成,由循环血液系统连接,每个隔室由组织体积(或重量)和组织血流速率定义,通常,这些隔室包括身体的主要组织,即脂肪组织、骨骼、大脑、肠道、心脏、肾脏、肝脏、肺、肌肉、皮肤和脾脏。然而,在某些情况下,也描述了简化模型,将具有相似血流率特性的组织合并在一起,以减少隔室数量和模型的整体复杂性。每个组织通常被描述为血流速率限制或渗透速率限制。
图2
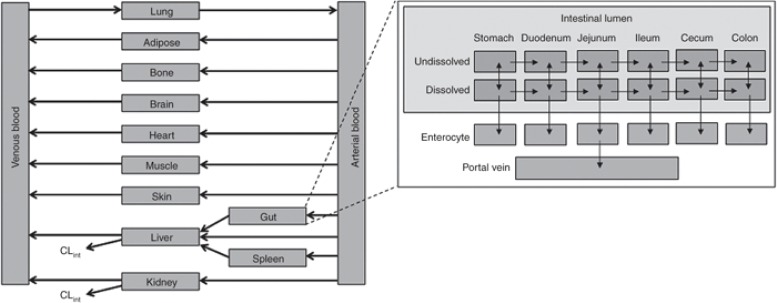 基于生理的药代动力学(PBPK)模型示意图
基于生理的药代动力学(PBPK)模型示意图
插图表示肠道的详细结构。CL int ,固有清除率;PBPK,基于生理的药代动力学。
图2
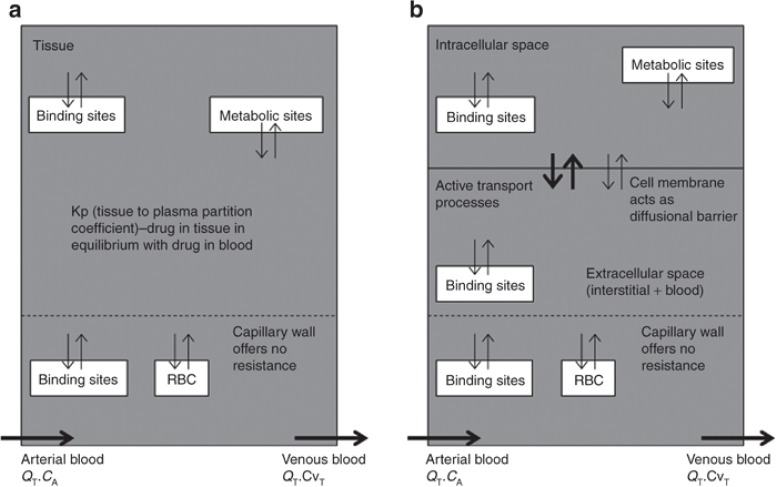
灌注与通透性限速组织模型。(a)灌注限速;(b)通透性限速。Kp,组织对血浆分配系数;RBC,红细胞。
血流灌注速率模型往往会发生在亲脂性小分子上,此时组织的血流成为限制过程。这种模型假定在稳态时组织中的总药物浓度与循环中的总药物浓度处于平衡状态,该平衡由特定药物的 Kp 值决定,而游离药物浓度(即未与蛋白质结合的药物浓度)相等。达到稳态所需的时间由血流速率、组织体积以及特定组织的 Kp 值决定。通常,高灌注的组织比灌注不良的组织更快达到稳态。渗透速率模型往往会发生在较大的极性分子上,此时细胞膜的渗透性成为限制过程。在这种情况下,组织基本上被分为两个部分,分别代表细胞内空间和细胞外空间,二者由作为扩散屏障的细胞膜隔开(图 2)。通常,在稳态下,该模型也会达到平衡,此时游离药物浓度通常相等。然而,对于此特定模型,达到平衡的时间高度依赖于药物特异性的渗透性,而非血流,血流用于估算驱动细胞膜两侧细胞内和细胞外浓度达到平衡的渗透率常数。如果涉及向细胞内或从细胞内进行的主动转运过程,细胞内的游离浓度可能高于或低于细胞外空间。此类主动转运过程通过将摄取参数纳入渗透率限制模型中进行建模。
药物研发中一般情况下生理药代动力学(PBPK)模型通常假定肝脏和肾脏是清除药物的唯一部位,并且遵循灌注速率限制的动力学规律。这些模型中使用的质量平衡微分方程已被多次描述,最近的一些描述可在 Jones 等人的出版物中找到。对于非消除组织,它们遵循以下原则,即“组织中药物的变化率”等于“流入率”(QT .CA)减去“流出率”(QT .CvT)。
![]()
Q = blood flow (l/h), C = concentration (mg/l), V = volume (l), T = tissues, A = arterial, v = venous, CvT = CT/(Kp/B:P), B:P = blood to plasma ratio.
对于组织消除而言,离开组织的静脉血中的游离浓度(假定其等于酶/消除部位的药物游离浓度)用于驱动清除率。在这种情况下,方程遵循以下原则,其中“流出速率”还包括消除速率(Q0 .Cv1 - CL2.Cv3)。
![]()
其中 CL int 为化合物的固有清除率(升/小时),u 表示未结合。CL int 指的是在没有蛋白质结合和血流等外在因素影响的情况下,化合物被相关酶代谢的内在能力(未结合)。
到目前为止所描述的模型可用于模拟静脉给药后的血浆和组织血浆浓度 - 时间曲线。对于口服给药,情况更为复杂,文献中已描述了多种吸收模型。从本质上讲,肠道(图 1)被分为两个主要隔室,分别代表肠腔(未吸收药物)和肠细胞(已吸收药物)。每个隔室进一步细分为多个子隔室,分别对应胃肠道的不同区域,即胃、十二指肠、空肠、回肠、盲肠和结肠。每个子隔室都由亚组织容积、转运时间和 pH 值来定义。亚组织容积和转运时间以类似于灌注组织方程的方式用于描述药物在胃肠道中的移动。在该模型中,使用特定于药物的参数(例如,离子化系数(pK),辛醇 - 水分配系数(logP)和溶解度)通过 pH 分配理论来描述药物在胃肠道腔内溶解和沉淀的过程。药物特异性渗透性数据用于模拟肠腔内溶解药物向肠细胞内吸收药物的吸收过程。通常,这些模型假设吸收是被动的,主动转运过程的贡献不大。然而,与灌注组织方程类似,如果能获得相关的药物特异性参数,也可以将主动摄取或外排转运过程纳入模型,分别用于驱动肠腔或肠细胞内浓度的模型估算。整合了分配(静脉注射)和口服吸收过程的 PBPK 模型已在文献中有所描述,并集成在商业平台中,包括 Simcyp(http://www.simcyp.com)、GastroPlus(http://www.simulations-plus.com)和 PKSIM(http://www.systems-biology.com/products/pk-sim.html)。另外,PBPK 模型也可以用常见的建模软件编写,例如 NONMEM、ADAPT、Berkeley Madonna、SAAM 和 WinNonlin 等,其示例可在在线补充数据中找到。
系统相关参数
已有物许多种开发了生理药代动力学(PBPK)模型,其中最常见的是小鼠、大鼠、狗和人类。 并且已被众多研究人员用于 PBPK 模型的构建。实际上,所有市售的 PBPK 软件都提供了最常见物种的 PBPK 模型。
鉴于这些模型的机械性本质,也可以将生理和机械特征纳入其中,以预测特定疾病状态和人群中的药代动力学(PK)和剂量。在此背景下,一些作者已将已知的肝血流量、细胞色素 P450(CYP)、肝体积、红细胞压积以及肝肾功能随疾病或年龄变化的情况纳入模型,以预测不同人群中的人体药代动力学。支持此类建模的数据库在文献中可以找到;这些数据库包括老年人群、肝肾功能受损者、儿科(包括发育阶段)、孕妇、肥胖人群、合并症如肝硬化和慢性肾衰竭患者,以及吸烟等环境因素。
生理药代动力学(PBPK)模型的一个关键优势在于能够将生理和生化变异性的来源纳入系统参数,并模拟个体群体而非平均个体的预期药代动力学。通过相关蒙特卡罗方法,可以利用描述人口统计学、解剖学和生理学变量的值和公式生成虚拟人群。PBPK 模型的系统参数分布方程是从基于真实人群和患者的分布数据中推导出来的。这使得在临床研究之前就能预测变异性,而统计方法(群体药代动力学分析)则需要先有临床数据来表征变异性。在考虑药物相互作用(DDI)相关风险时,能够评估人群中的变异性尤为重要,因为通常只有少数具有特定特征的个体比平均个体更令人担忧。
药物特异性参数
使用通用的生理药代动力学(PBPK)模型来模拟静脉注射和口服给药的血浆浓度-时间曲线时,还需要额外的药物特异性输入参数(例如,CL int 和 Kp 值)。了解特定化合物的关键吸收、分布、代谢和排泄(ADME)机制以及明确且可测量的药物特异性参数是预测成功的关键。体内器官的内在清除率是 PBPK 模型中表征体内清除率的关键参数。对于肝清除率,可以从各种体外系统(例如,重组酶、微粒体和肝细胞)通过生理缩放因子(如系统间外推因子、微粒体回收率、肝细胞数)进行缩放。对于微粒体和肝细胞,这些缩放计算分别在公式 3 和公式 4 中给出。
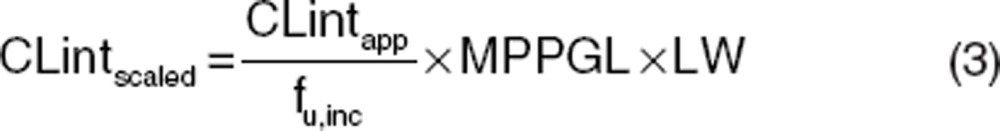
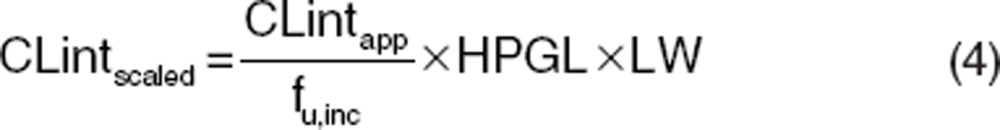
CLint scaled 是体内校正后的清除率(单位为毫升/分钟),CLint app 是体外表观清除率(对于微粒体单位为微升/分钟/毫克,对于肝细胞单位为微升/分钟/百万个细胞),f u,inc 是体外系统中的未结合分数,MPPGL 是每克肝脏的微粒体蛋白毫克数(毫克/克),LW 是肝脏重量(千克),HPGL 是每克肝脏的肝细胞数(10 6 /克)。
这些值在转换为相关单位后可直接用于生理药代动力学(PBPK)模型,或者与外在因素(例如血浆蛋白结合数据和肝血流量)一起用于定义明确的肝模型(如全混流模型和平行管模型)来预测肝清除率。许多作者已经详细描述了这些预测肝清除率的方法,并且这些方法已经得到了广泛的验证。对于其他清除机制,如肾/胆汁排泄,可以使用多种方法来预测体内器官的内在清除率。最常用的是基于异速生长的方法,其形式如方程 5 所示,其中未结合的清除率是从单个物种或多个物种进行缩放的。

其中 a 是异速生长系数,b 是异速生长幂函数,BW 是体重。这些预测的清除率值需要在输入 PBPK 模型之前转换为相关组织中的体内 CL 值。或者,对于某些机制(例如胆汁排泄),有体外 - 体内缩放方法可用,并且已在少数情况下使用。
另一组重要的化合物特异性参数是 Kp 值,用于表征化合物在体内不同组织中的分布或“移动”。Kp 值被定义为稳态时组织中化合物的总浓度与血浆中化合物总浓度的比值。从更机理的角度来看,这些 Kp 值反映了由于蛋白质结合、溶酶体捕获和脂质溶解等过程而导致的组织蓄积程度。过去,通常通过成本高昂且耗时的临床前组织分布研究来实验测定 Kp 值,在这些研究中,会随时间推移并在稳态时测量组织和血浆中的药物浓度。然而,近年来,文献中已描述了多种用于预测 Kp 值的机理方法。基于组织成分的机理方程的发展,用于预测大鼠、狗和人类的 Kp 值以及分布情况,彻底改变了并更全面地推动了早期药物发现中常规应用 PBPK 方法。这些模型并非需要体内数据,而是根据化合物的物理化学性质以及与脂质和蛋白质的体外结合特性来估算其在组织中的分布程度。普林及其同事开发的方程假定药物通过被动扩散均匀分布到组织和血浆中,其中非特异性结合到脂质的程度由药物的亲脂性数据估算,而与血浆和组织中蛋白质的特异性可逆结合程度则由血浆蛋白结合估算得出。罗杰斯及其同事 通过引入离子化/电荷考虑因素对这些方程进行了扩展。这些方程考虑了未电离药物向中性脂质和中性磷脂的分配、电离和未电离药物在组织水中的溶解、强电离碱与酸性磷脂之间的静电相互作用,以及中性、弱碱和酸与细胞外蛋白的相互作用。上述基于组织成分的方程各自旨在描述特定的相互作用。近来,已开发出将这些不同的单独机制统一起来的算法,以方便其应用。此外,V 可以使用公式 6 进行计算。
![]()
VP = volume of plasma
已开展多项研究,旨在利用一系列药物数据集来探究这些不同机制方法对 Kp 预测的可预测性,其准确度各不相同。需要注意的是,使用基于计算机的 Kp 参数(即没有实测测量组织数据)构建的 PBPK 模型可能对组织动力学的描述过于简化。如果对特定目标组织中的药物浓度特别感兴趣,则可能需要体内组织分布数据来进一步完善模型。
一个重要组成部分是任何口服药代动力学模拟的预测吸收速率和吸收程度。Simcyp(“高级溶出、吸收和代谢”模型;ADAM)、GastroPlus(“高级隔室吸收转运”模型;ACAT)和PKSIM开发的吸收模型已在文献中详细描述。此类软件依靠各种体外和/或计算机模拟输入数据,如溶解度、渗透性、粒径、logP和pK值,来模拟化合物在消化道不同部位转运过程中与溶出、沉淀、摄取和吸收相关的动力学。
吸收的一个关键输入参数是对人体有效渗透性的度量。在药物发现的早期阶段,这可以通过计算机模拟模型预测,或者通过高通量检测方法来测定。在更高级的阶段,通常更倾向于使用 Caco-2 细胞系的渗透性测量值。要在生理药代动力学模型中利用此类数据,必须将这些体外数据按体内情况(人体有效渗透性)进行缩放。测试化合物通常会与若干已测得人体在体空肠渗透性数据的参考药物进行校准。
另一个关键的输入参数是体内相关的溶解度。在给定的 pH 值下,该值可用于利用化合物特定的 pKₐ 和 pH 分配理论来估算胃肠道内一系列 pH 值下的溶解度。对于高溶解度的化合物,水溶性数据可预测体内情况,并且可以放心地用于 PBPK 模型。然而,对于溶解度差且亲脂性强的化合物,水溶性值往往低估了体内溶解速率,因为胆汁盐和脂质可以增强溶解。在这种情况下,应生成更多在诸如空腹状态模拟肠液和进食状态模拟肠液等生物相关介质中的溶解度数据。许多作者已经表明,这些溶解度测量对于动物和人类可靠的 PBPK 模拟至关重要。
生理药代动力学(PBPK)模型提供了一个生理学框架,便于根据需要纳入其他机制,例如主动转运过程。如果相关输入数据可用,此类转运过程可在多个不同组织中纳入,例如肝脏和肠道,与血浆相比,可能会导致药物游离浓度升高或降低。对于肠道而言,可通过在一系列化合物浓度下对测试化合物进行孵育,从 Caco-2 细胞系统中获得描述药物外排动力学的参数。这些参数可通过校正体外与体内表面积差异进行缩放,并在 PBPK 模型中使用,以模拟 P-糖蛋白对化合物吸收的影响。然而,此类应用目前受限于 P-糖蛋白动力学参数的体外-体内相关性缺乏,主要依赖于“模型拟合”而非“模型模拟”。对于肝脏而言,有机阴离子转运蛋白(OATP)介导的摄取也可纳入通用 PBPK 模型框架。这是通过将肝脏建模为具有渗透限制的组织来实现的,该模型在窦状膜处纳入了未结合药物的主动摄取和被动扩散,在胆小管膜处纳入了未结合药物的胆汁排泄。支持这些模型的化合物特异性参数可以从体外夹层培养的肝细胞数据中估算,并根据先前所述的每克肝脏的肝细胞数和肝脏重量进行调整以适应体内情况。夹层培养的肝细胞实验通常测量在存在和不存在抑制剂/条件以促进主动摄取和排泄的情况下,化合物在肝细胞中的“量”随时间的增加。为了准确确定这些转运参数,特别是胆汁排泄参数,必须估算细胞内化合物的浓度,因此已采用描述肝细胞系统动力学的体外模型来精确计算这些体外摄取参数。将相关的体外参数按比例整合到生理药代动力学(PBPK)模型中,已被用于模拟大鼠和人类有机阳离子转运多肽(OATP)底物的体内药代动力学(PK)。在大多数情况下,只有在引入经验比例因子时,才能成功做出预测。
药物发现与开发中的生理药代动力学建模策略
生理药代动力学(PBPK)模型可用于药物发现和开发的早期阶段,即先导化合物开发之前,此时可用数据有限,也可用于药物开发的早期至晚期阶段,此时可用数据较多。目前已有多个实例表明,在药物发现和开发阶段使用 PBPK 模型有助于做出与候选药物选择、首次人体剂量、药物相互作用(DDI)潜力评估以及涉及 DDI 的适当研究设计或使用多态酶代谢药物的研究的纳入/排除标准相关的决策。在整个药物发现和开发过程中,随着来自临床前和临床研究的有关药物分布和吸收的更多信息的出现,PBPK 模型可以进行迭代改进。构建良好的 PBPK 模型在临床前和临床药理学研究的设计中可发挥重要作用。在此,我们介绍了 PBPK 模型应用的策略。Jones 等人提出了用于模拟新化学实体的人体药代动力学的 PBPK 模型应用策略,并对其进行了验证。
图3
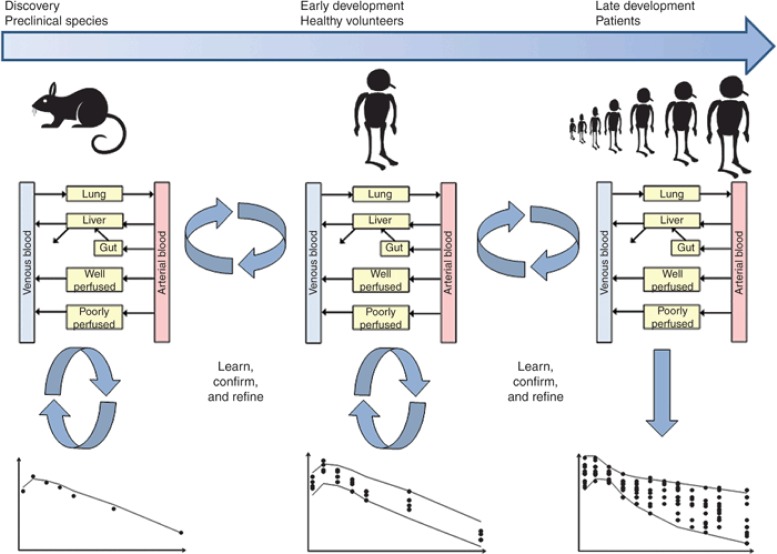
基于生理学的药代动力学(PBPK)模型在药物研发中的策略。推荐采用一种迭代的“学习、确认和优化”的 PBPK 模拟方法。最初,在动物身上使用动物 PBPK 模型、动物体外数据以及化合物特定的物理化学数据进行 PBPK 模拟。将动物模拟结果与体内数据进行比较,如果动物模拟合理,则使用基于健康志愿者生理学、人体体外数据以及化合物特定物理化学数据构建的人体 PBPK 模型对健康志愿者进行模拟。然后,可以使用相关生理学将这些模拟扩展到各种患者群体。如果在任何阶段模拟结果不准确,这表明违反了一个或多个模型假设,在这种情况下,可能需要进行进一步的实验以了解不匹配的原因。PBPK,基于生理学的药代动力学。
最初,使用动物的生理药代动力学(PBPK)模型、动物体外数据以及化合物特定的物理化学数据进行模拟。然后将模拟结果与实际的体内数据进行比较。这一步骤的目的是验证模型对于目标化合物的假设是否成立。如果模拟曲线能够重现体内数据(通过目测检查),则使用人体 PBPK 模型、人体体外数据以及化合物特定的物理化学数据进行模拟。如果动物模拟结果与体内数据不一致,这可能表明影响药物药代动力学的某种生物学机制未被纳入模型,且不太可能通过 ADME 筛选试验来体现。在这种情况下,PBPK 模型可用于推测模拟结果与实际体内数据不匹配的原因,并可能指导进一步实验的设计。这种方法已由多个独立小组使用不同的药物数据集进行了验证。在文献中可以找到使用 PBPK 模型成功预测人体药代动力学的具体实例。这些出版物提供了药代动力学模型在首次人体临床试验中应用的实例。
一旦利用临床数据对这种人体模型进行了验证,就可以前瞻性地将其应用于评估不同给药方案的影响,或者评估化合物作为底物或诱导剂的药物相互作用(DDI)潜力。随后,可以将预测结果与实际数据进行比较,一旦发现任何不一致之处,就可以对临床数据和体外数据进行审查,以确定模型中是否存在缺失的成分(例如,可能导致剂量非线性的自身抑制)或体外参数是否存在错误(例如,对清除率的低估导致对清除率的预测不足)。尽管体外数据为模型应包含的关键成分提供了指示,但临床数据也是信息来源。例如,单次递增剂量数据和多次递增剂量数据可以表明剂量依赖性或时间依赖性动力学是否存在问题。此外,临床 DDI 数据可用于评估底物药物的 f1 值(由受抑制酶代谢的分数)的稳健性,这是预测 DDI 的重要参数。一旦确定了“缺失的成分”或“错误的参数”(参数 X),就可以将“自上而下”的拟合方法与“自下而上”的方法相结合,以获得参数 X 的估计值。临床数据(例如血浆浓度 - 时间曲线)可以与所有先前的体外数据的体外到体内推算(IVIVE)相结合,使用最小二乘法拟合算法反复拟合参数 X,直到模拟的浓度 - 时间曲线与临床数据一致。然后对 PBPK 模型进行验证,以确保包含参数 X 后能够恢复观察到的数据。后者应来自独立验证集,即未用于原始模型开发的临床数据。如果模型无法恢复观察到的数据,则应相应地对模型进行修订。这一迭代过程在 Vieira 等人的文章中有所描述,并在图 3 中有所展示。这可能更适用于药物开发的后期阶段,在此阶段的最终目标是开发一个能够恢复所有临床场景的 PBPK 模型,从而确保所有相关的机制成分都已整合。
尽管在将转运蛋白纳入生理药代动力学(PBPK)模型以及预测转运蛋白介导的药物相互作用(DDIs)方面已取得近期进展,但仍有许多挑战有待阐明。例如,体外系统存在一些问题,且在模拟体内情况时存在生理学上的局限性。这包括酶与转运蛋白之间的相互作用,以及当一种转运蛋白的活性受到抑制时,其他一种或多种转运蛋白活性可能代偿性增加的情况。近期的出版物表明,当化合物的处置涉及转运蛋白介导的摄取时,需要采用“自上而下/自下而上”的拟合方法,以恢复临床数据。
为了促进和加快模型构建,如果有多名受试者的临床数据可用,就可以使用群体药代动力学分析,包括最大似然法或贝叶斯方法,来获得参数的最佳估计值。贝叶斯方法通过纳入各种未知参数的先验分布(包括体外数据或临床试验数据)来扩展最大似然法。因此,鉴于贝叶斯方法纳入了大量先验信息,它已成为生理药代动力学模型的最佳选择。
制药公司内部许多人的观点似乎是,如果利用一些临床数据开发并验证了生理药代动力学(PBPK)模型,那么就可以前瞻性地将其用于预测药物相互作用(DDIs),最终目的是让监管机构豁免临床 DDI 研究。这种方法存在的一个问题是,这又引出了一个问题,即该模型的可靠性如何?当然,这又成了一个循环论证,因为没有临床 DDI 数据来支持模型的稳健性。必须进行临床 DDI 研究并不否定 PBPK 模型在药物开发过程中的影响;它只是增强了模型的作用。如前所述,这些数据可用于确保 PBPK 模型的稳健性。经过改进和验证的模型随后可用于前瞻性地预测处于极高风险的个体或出于伦理原因无法在正式临床试验中进行研究的受试者的药代动力学和药物相互作用。
在生理药代动力学(PBPK)模型中构建“系统”属性,能够对诸如种族、遗传、年龄、肝病、肾功能损害和发育阶段等协变量对生理参数(如细胞色素 P450 酶丰度、每克肝脏的微粒体蛋白以及肝脏体积)的影响进行定量评估。因此,一旦在健康志愿者群体中开发并验证了 PBPK 模型,就可以使用相关的“系统”参数在目标人群中进行模拟(图 3)。如果针对感兴趣的人群没有现成的数据库,只要已知疾病的病因,就可以构建一个。鉴于监管机构显然已接受这种方法,用于预测特殊人群药代动力学暴露的 PBPK 模型的使用正在增加。实际上,目前针对肝功能损害患者的药代动力学监管指南建议开发 PBPK 模型。此外,要求在完成 I 期试验前提交儿科研究计划,这促使许多公司使用 PBPK 技术来预测儿童的药代动力学并确定剂量。
生理药代动力学模型的应用实例
示例 1:发现与早期开发阶段
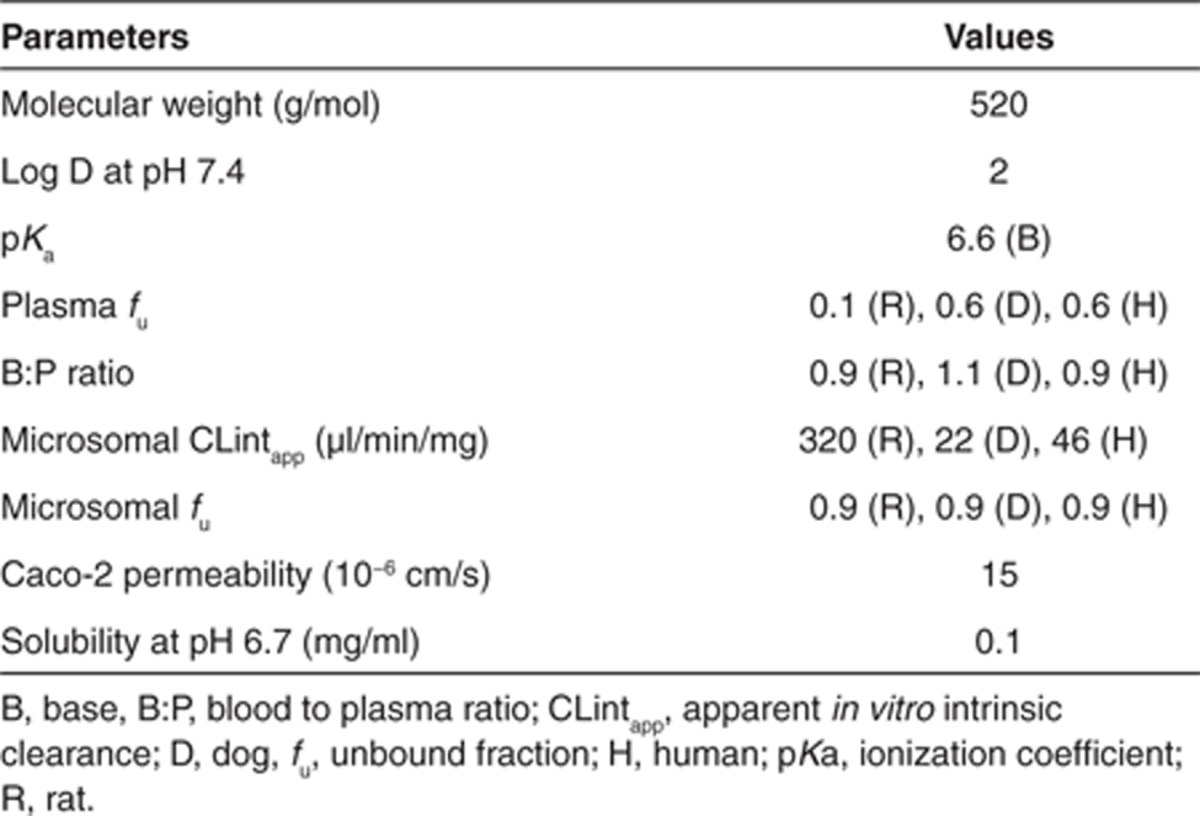
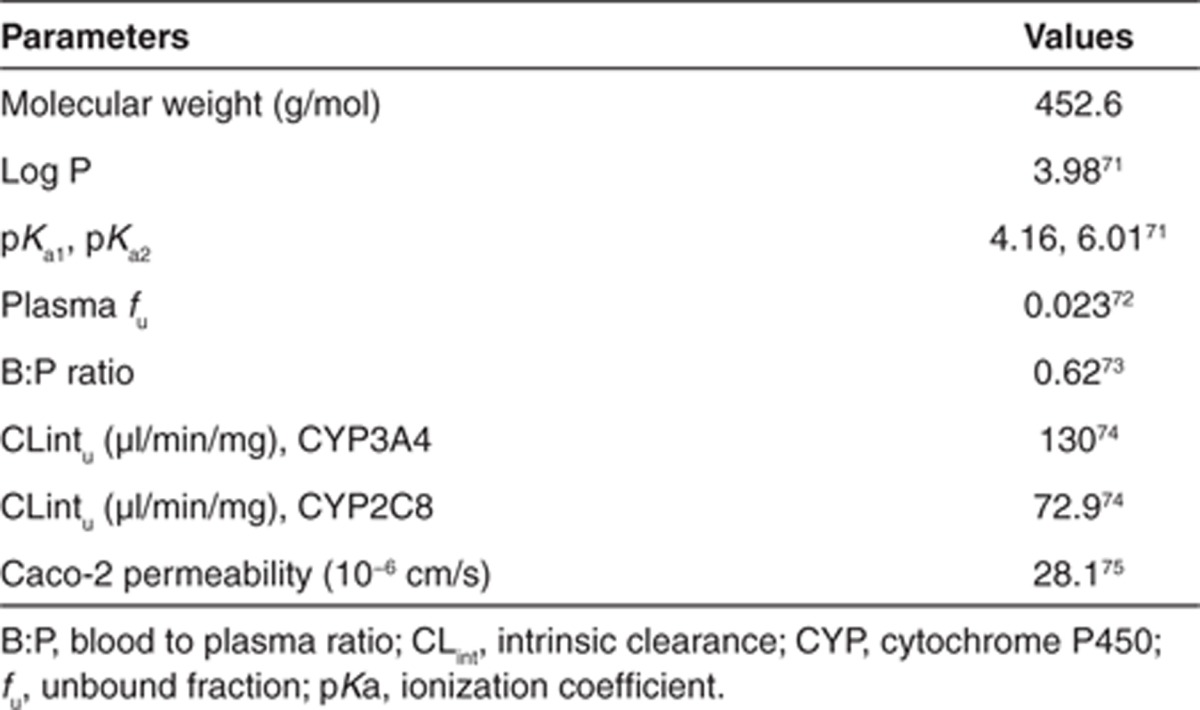
这里描述了一个使用生理药代动力学(PBPK)模型对人类药代动力学进行前瞻性预测的“真实生活”示例。化合物 X 是一种中等亲脂性、弱碱性物质,具有良好的渗透性和中等溶解度。临床前的体外和体内研究表明,化合物 X 主要通过 CYP450 途径清除。其可用的物理化学性质和体外特性见表 1。
表 1. 化合物 X 的 PBPK 特定输入参数。
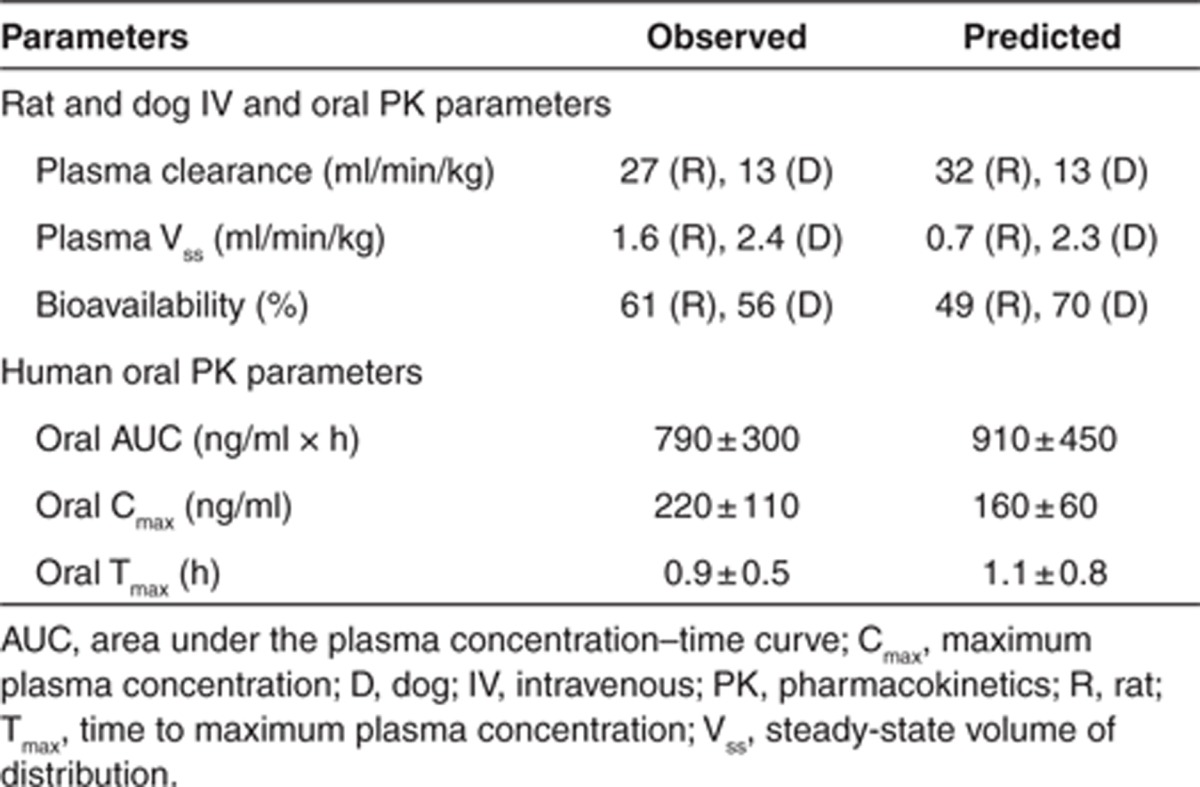
基于生理的药代动力学(PBPK)模型及假设最初在大鼠和犬体内通过静脉注射和口服给药进行了验证。如图 4a、b 所示,大鼠和犬的静脉血浆浓度 - 时间曲线由 PBPK 模型预测得相当准确,这表明 CYP 代谢(分别根据大鼠和犬的肝微粒体预测)以及被动、灌注限制性分布(使用根据组织成分方程预测的 Kp 值)的假设是合理的。此外,预测的药代动力学参数(清除率 CL 和分布容积 V)与观察到的数据非常吻合(表 2)。
图4
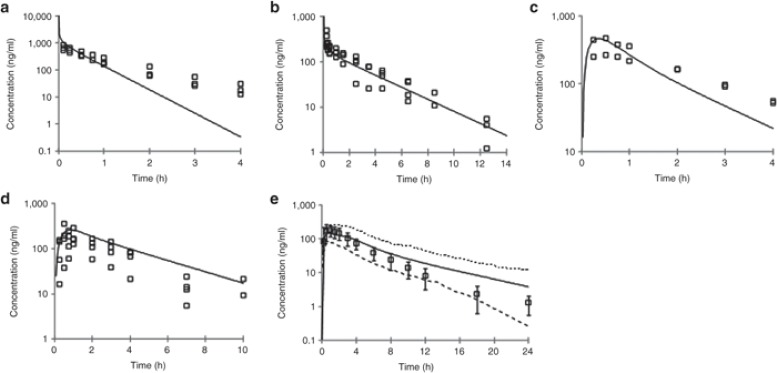
大鼠、犬和人的血浆浓度 - 时间曲线的观察值与预测值。(a)大鼠静脉注射(1 毫克/千克);(b)犬静脉注射(0.5 毫克/千克);(c)大鼠口服(2 毫克/千克);(d)犬口服(1 毫克/千克);(e)人口服(100 毫克)。使用 SimCYP 基于人群的模拟器进行模拟。在 a - d 中:空心方块 = 观察数据;实线 = 模型预测;在 e 中:空心方块 = 平均观察数据 ± 标准差;实线 = 平均模型预测;虚线 = 模型预测的 5
表 2. 化合物 X 的观察到的与预测的药代动力学参数。
为了评估模型中吸收部分的有效性及其假设,对静脉注射模型进行了优化,使其与观察到的静脉注射数据和 Caco-2 通透性数据精确匹配,并使用水溶性数据来模拟吸收。在此情况下,PBPK 模型能够准确地捕捉到两种物种中观察到的口服血浆浓度 - 时间曲线(图 4c、d),表明该化合物的被动吸收假设是有效的。此外,利用预测的吸收和清除率,生物利用度得到了准确的估计。在进行人体模拟之前,这种在大鼠和狗身上进行的初步验证为模型假设和预测能力提供了信心。
在人体中以建议的有效剂量100 毫克进行了模拟实验,采用了与大鼠和狗实验相同的假设,即 CYP450 介导的代谢(根据人肝微粒体预测)、被动、灌注限制性分布(使用根据组织成分方程预测的 Kp 值);以及使用 ADAM 模型的被动吸收(以 Caco-2 和溶解度数据作为输入)。如图 4e 所示,模拟的口服血浆浓度 - 时间曲线(平均值和 5
示例 2:临床开发阶段
在此,我们描述了一种方法,该方法可用于临床和体外数据均可用,但对 PBPK 模型中某一特定参数存在不确定性的情况。瑞格列奈是一种短效的格列奈类降糖药,用于治疗 2 型糖尿病。口服给药后,该药物迅速吸收,并经历首过代谢,生物利用度为 60
表 3. 预混胰岛素瑞格列奈的 PBPK 特定输入参数。
这些数据被用于驱动 PBPK 模型,假设在肝脏中分布受渗透性限制。将 Niemi 等人报告的研究中瑞格列奈的“自上而下”拟合方法(平均浓度-时间曲线) 67 与所有先前体外数据的“自下而上”外推相结合,以获得瑞格列奈通过 OATP1B1 介导的肝摄取 CL int 的估计值为 282 μl/min/百万细胞。包括通过 OATP1B1 的肝摄取在内的模拟能够恢复不同剂量下的观察数据(步骤 A;图 5)。为了确定 CYP2C8、CYP3A4 和 OATP1B1 对瑞格列奈处置的相对贡献是否恰当,使用抑制剂甲氧苄啶(CYP2C8)、克拉维霉素(CYP3A4)和环孢素(OATP1B1)模拟药物相互作用,并与观察数据进行比较。在此之前,运行了每种抑制剂的模拟浓度-时间曲线,以确保每种抑制剂的 PBPK 模型能够恢复观察到的曲线(步骤 B;图 5)。此外,这些复合文件经过验证,以确保其能够准确预测与包括咪达唑仑(CYP3A4)、罗格列酮(CYP2C8)和瑞舒伐他汀(OATP1B1)在内的探针底物的药物相互作用。因此,即使在研究单一药物相互作用对时,也应研究相互作用矩阵,以确保 PBPK 模型的所有组成部分都具有稳健性。平均而言,在与甲氧苄啶(1.3 倍;160 毫克/天)、克拉维酸(1.4 倍;250 毫克/天)和环孢素(两倍;100 毫克/天)联合给药期间,瑞格列奈血浆浓度-时间曲线下面积的预测增加值分别为 1.6 倍、1.4 倍和 2.4 倍,与观察到的值一致(步骤 C;图 5)。尽管此处仅展示了血浆浓度的变化,但完整的 PBPK 模型可用于预测药物相互作用对瑞格列奈在胰腺(在此情况下为作用部位)暴露的影响。然后,模拟的胰腺浓度可用于驱动响应,该响应可通过应用 PD 模型进行研究。
图 5
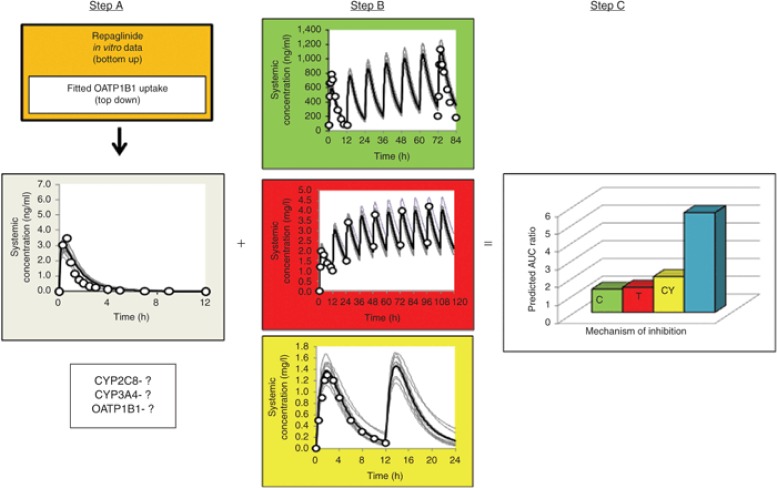
用于预测药物相互作用的生理药代动力学建模策略。C(绿色)=克拉霉素;T(红色)=甲氧苄啶;CY(黄色)=环孢素。步骤 A 指的是通过“自上而下/自下而上”的组合方法开发初始底物模型。然后需要通过模拟和已知药物相互作用研究的恢复来评估模型的准确性,以确认 CYP 和 OATP 组分的相对贡献。步骤 B 指的是在模拟药物相互作用之前对抑制剂模型进行验证,通过比较模拟和观察到的曲线来进行。步骤 C 指的是使用经过验证的底物和抑制剂模型来模拟药物相互作用,以确认最终的底物模型能够恢复观察到的药物相互作用。AUC,血浆浓度-时间曲线下面积;CYP,细胞色素 P450;DDI,药物相互作用;OATP,有机阴离子转运蛋白;PBPK,基于生理的药代动力学。
需要将转运蛋白和 CYP 酶以及其他内在和外在患者因素纳入的生理药代动力学(PBPK)模型,来评估个体发生药物相互作用(DDI)的风险,尤其是涉及多种抑制剂的情况。这对监管机构来说尤其重要。不可能对所有可能的组合进行体内研究。模拟可用于评估临床评估中的最坏情况组合。这很可能涉及抑制不同酶或转运蛋白的药物组合,其效果是累加的,如此处所示的例子。在我们的虚拟临床试验中,三种抑制剂与瑞格列奈同时给药,平均血浆浓度-时间曲线下面积增加了 5.6 倍(图 5),但在虚拟人群中,该值范围为 2.3 至 18 倍。尽管没有临床数据来证实涉及抑制 CYP2C8、CYP3A4 和 OATP1B1 介导摄取的复杂药物相互作用的预测强度是否准确,但 PBPK 模型能够恢复涉及单一抑制剂的每个临床药物相互作用的观察数据这一事实,为其准确性提供了一定的信心。
未来的视角
尽管令人欣慰地看到,PBPK 模型在制药行业中越来越多地用于预测药代动力学,但显然仍存在一些重大挑战需要解决,以提高这种方法的成功率。许多“系统参数”仍缺乏,比如酶、转运蛋白和其他相关蛋白质的丰度。不同种族人群和疾病组的生理学和生物学信息也很匮乏。尽管如此,人们还是更侧重于改进体外系统及相关方法,以准确预测药物的 ADME 参数。对于低清除率化合物而言,情况尤其如此,因为目前的技术难以测量人体肝组织中的药物消耗率。随着人们努力开发代谢更稳定的化合物,由转运蛋白介导的药代动力学变得越来越普遍。尽管在将转运蛋白纳入 PBPK 模型方面已取得了一些进展,但人们已经认识到体外系统存在一些问题,以及在模拟体内情况方面存在生理学限制,还需要做更多的工作。制药行业内部的经济限制导致了预竞争研究合作的增长,这或许有助于解决此类问题,同时也会增加“系统参数”的可用性,因为资源问题可以在相关方之间分担。
未来的发展包括将基于生理的药代动力学(PBPK)模型与完全机制性的药效学(PD)模型相结合,并考虑药理反应的变异性(包括受体基因型)。此外,系统药理学可能会被视为药代动力学/药效学(PK/PD)的下一个前沿领域,在这个领域中,从剂量到暴露(PBPK 模型)再到反应(PD 模型)的每一步都可以纳入机制细节。这或许可以被视为迈向“个性化医疗”的第一步。实际上,在未来的某个阶段,或许有可能根据已知的与药物吸收、分布、代谢和排泄(ADME)以及药理学相关的蛋白质基因型,预测特定个体的药物药代动力学、疗效和副作用。另一个发展包括将 PBPK 模型常规应用于预测大分子的分布情况。商业平台中集成的 PBPK 模型在制药行业中得到应用,这些平台不断更新以纳入这些科学进展。因此,用户必须接受足够的科学知识教育以及软件功能培训。这可以通过研讨会的形式提供,甚至可以在学术环境中以相关学科化的方式进行教学。话虽如此,经验才是最重要的。在制药行业,应指派专门的用户来开发和保持应用 PBPK 模型所需的技能组合。由于 PBPK 建模具有多学科的特点,用户往往具有各种各样的背景,包括药物代谢、药理学、医学、药学、生物物理学、工程学、数学、编程和统计学。因此,促进跨学科的交流非常重要。
结论
毫无疑问,利用生理药代动力学(PBPK)模型来最大化药物的临床潜力已被制药行业和监管机构所接受,因此,其应用范围可能会更广。PBPK 模型已被认为是复杂且数据密集型的。随着我们对生理学和生化过程的了解不断加深,尤其是在不同疾病状态下,将会开发出更复杂的模型。因此,无论用户的专业水平如何,随着模型的发展,都需要持续的教育。在药物开发中使用 PBPK 模型需要充足的资源,个人也需要在模型应用方面接受足够的培训,并且对驱动模型所需的吸收、分布、代谢和排泄(ADME)数据有良好的理解。在临床前药物发现和临床药物开发人员之间保持良好的沟通,有助于提供可靠的 PBPK 模型,然后可以前瞻性地应用于回答许多与药物开发过程相关的问题。


文章评论