摘要
Esaxerenone 是一种口服非甾体选择性盐皮质激素受体阻断剂。本文整理 Yamada 等发表于 Drug Metabolism and Disposition 的研究,重点关注其作为相互作用诱发药(perpetrator)时,是否会影响其他药物的暴露。研究显示,Esaxerenone 在体外同时表现出 CYP3A 时间依赖性抑制和 CYP3A4 诱导作用。若分别评估抑制和诱导效应,midazolam 暴露变化提示 DDI 风险不能忽视;但在 PBPK 模型中联合纳入两类机制后,预测的 midazolam AUCR 约为 1.2,与临床结果接近。总体来看,Esaxerenone 虽存在 CYP3A 相互作用信号,但由于抑制与诱导在体内发生部分抵消,其作为 perpetrator 的临床 DDI 风险较低。
引言
药物相互作用风险评估是新药研发中的重要内容。对于一个新药,除了关注它作为受体药(victim)时会不会被别的药影响,更值得问的另一个问题是:它作为相互作用诱发药(perpetrator)时,会不会影响别的药。
这篇文章的重点就在这里。体外实验提示,Esaxerenone 对 CYP3A 并不是单一方向的作用,而是同时具有抑制和诱导潜能。对于这类“既有抑制又有诱导”的化合物,如果只用传统静态模型评估,往往容易得到偏保守的结论;而 PBPK 模型由于能整合体内动态暴露,更适合用来判断真实临床风险。因此,这篇文章的核心并不只是报告体外结果,而是通过体外实验、静态模型和 PBPK 模型逐步评估 Esaxerenone 作为 perpetrator 时的实际 DDI 风险。
研究方法
作者首先在体外系统评估了 Esaxerenone 对多个代谢酶和转运体的影响。CYP 抑制实验采用 pooled human liver microsomes,覆盖 CYP1A2、2A6、2B6、2C8、2C9、2C19、2D6、2E1 和 3A4;UGT 部分评估了 UGT1A1 和 UGT2B7。其中,CYP3A4 使用了 midazolam 和 testosterone 两种探针底物。若在有无 NADPH 预孵育条件下观察到抑制增强,则进一步计算 kinact 和 KI,用于判断是否存在时间依赖性抑制(TDI)。
诱导实验采用原代人肝细胞培养体系,分别评估 CYP1A2、CYP2B6 和 CYP3A4 的酶活和 mRNA 水平变化。若出现浓度依赖性诱导,则进一步拟合 Emax 和 EC50。与此同时,作者还检测了多个关键转运体,包括 OAT1、OAT3、OATP1B1、OATP1B3、OCT1、OCT2、MATE1、MATE2-K、P-gp 以及 BCRP,并补充测定了 Esaxerenone 在血浆和微粒体中的蛋白结合率以及血浆/全血分配比,为后续模型输入提供参数。
在静态模型部分,作者依据 FDA DDI 指南,使用 mechanistic static model 评估 Esaxerenone 对 CYP3A 底物和 CYP2B6 底物的影响,并以 midazolam 作为典型 CYP3A 底物。模型中分别计算了肠道和肝脏中的抑制项、TDI 项和诱导项,同时给出 AUCRinh、AUCRind 和 AUCRtot。其中,midazolam 的 Fg 取 0.54,fm 取 0.94,肠道与肝脏中的作用浓度则分别由吸收参数、Cmax、fu,p 和血流参数估算。
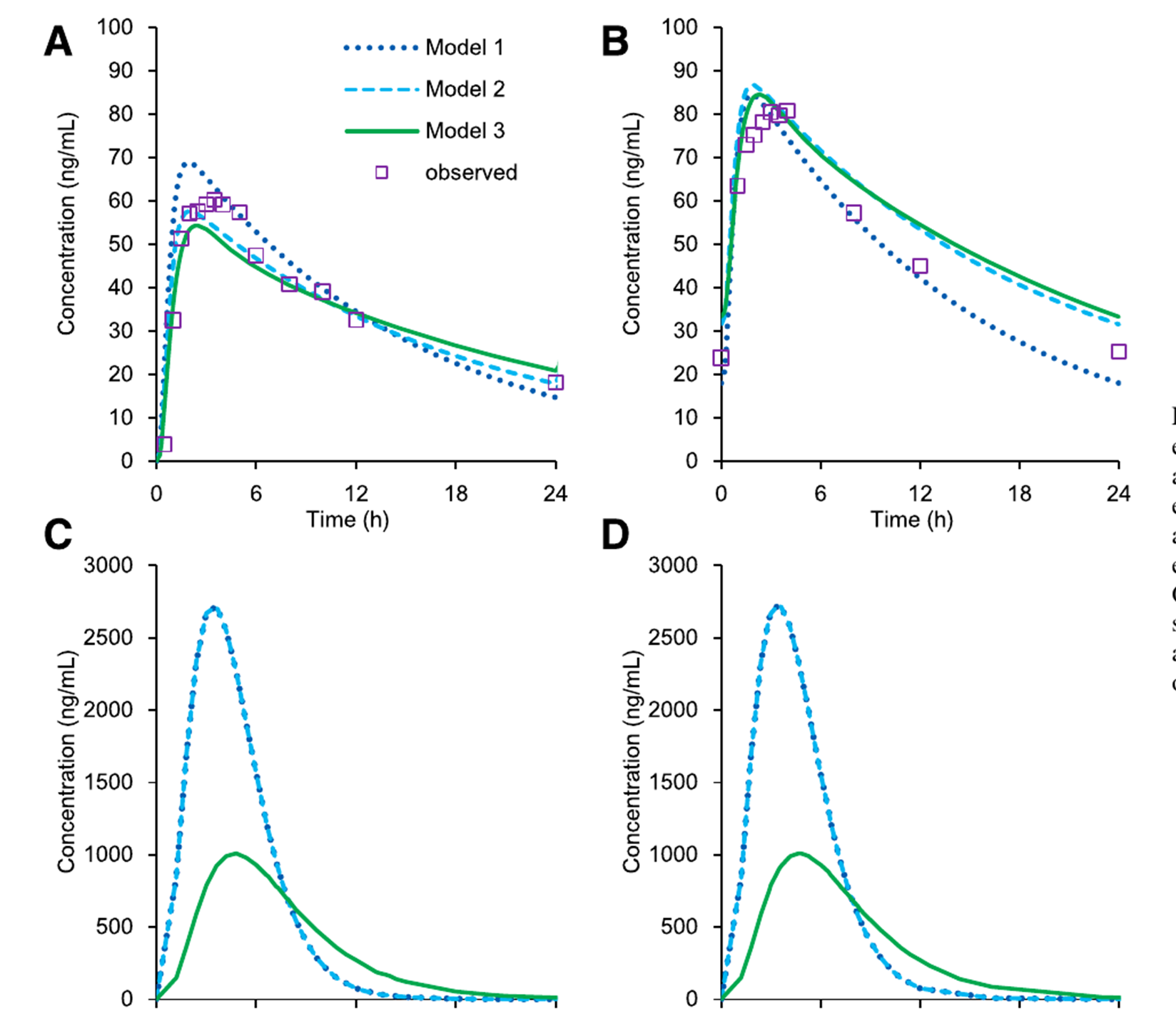
在 PBPK 部分,作者使用 GastroPlus 9.7 建立 Esaxerenone 模型,并调用既往建立的 midazolam 模型进行 DDI 模拟。文中一共构建了三套人体 PK 模型:模型 1 基于猴静脉给药数据通过单物种异速生长外推;模型 2 基于口服临床数据优化拟合;模型 3 结合静脉和口服临床数据构建。作者不仅比较了三套模型在临床给药条件下对 midazolam AUCR 的预测,还进一步模拟了 不同给药天数、不同给药时点以及不同 Esaxerenone 剂量 对 DDI 的影响。
体外结果
整体来看,Esaxerenone 对多数酶和转运体的抑制作用较弱,临床相关性有限。真正需要重点关注的是 CYP3A。作者观察到 Esaxerenone 同时存在 可逆抑制、时间依赖性抑制(TDI)和 CYP3A4 诱导。使用 midazolam 作为底物时,CYP3A4 的 Ki 约为 40.3 μM,kinact 约为 0.0235 min-1,KI 约为 44.8 μM。在原代肝细胞中,CYP3A4 诱导参数均值为 Emax 37.4,EC50 10.5 μM。
也就是说,Esaxerenone 对 CYP3A 的影响是双向的:一方面它会降低酶活性,另一方面又会提高酶表达。对于这种同时具备抑制与诱导潜力的化合物,后续判断不能只停留在体外信号层面,而必须进一步看两种机制在体内的净效应。
静态模型结果
在 mechanistic static model 中,如果将抑制和诱导分开评估,Esaxerenone 显示出不能忽视的 DDI 信号。对于 midazolam,仅考虑抑制时 AUCR 约为 1.80,仅考虑诱导时 AUCR 约为 0.31。这两个结果都说明,如果只抓住某一侧机制,很容易得出“风险较明显”的判断。
当在静态模型中同时纳入抑制和诱导后,midazolam 的 总 AUCR 仍约为 1.30。不过,这一结果本身也反映出静态模型的特点:它更适合作为保守筛查工具,往往容易高估真实体内风险,尤其是在药物同时存在多种方向相反机制时。
PBPK 模型结果
PBPK 模拟结果比静态模型更接近真实临床观察。尽管三套 Esaxerenone 模型对血浆浓度的拟合并不完全相同,但对肠上皮细胞浓度(Cent)的预测较为接近,因此对 midazolam AUCR 的预测也较一致。在联合纳入抑制与诱导机制后,三套模型预测的 midazolam 总 AUCR 约为 1.17-1.23,与临床研究观察到的约 1.2 很接近。
作者进一步分解肠道和肝脏贡献后发现,AUCRh 基本接近 1,提示相互作用主要发生在肠道,而不是肝脏。换句话说,对于 Esaxerenone 这种弱 CYP3A perpetrator,肠道局部暴露比系统循环暴露更关键。这也是 PBPK 模型在本文中比静态模型更有解释力的重要原因。
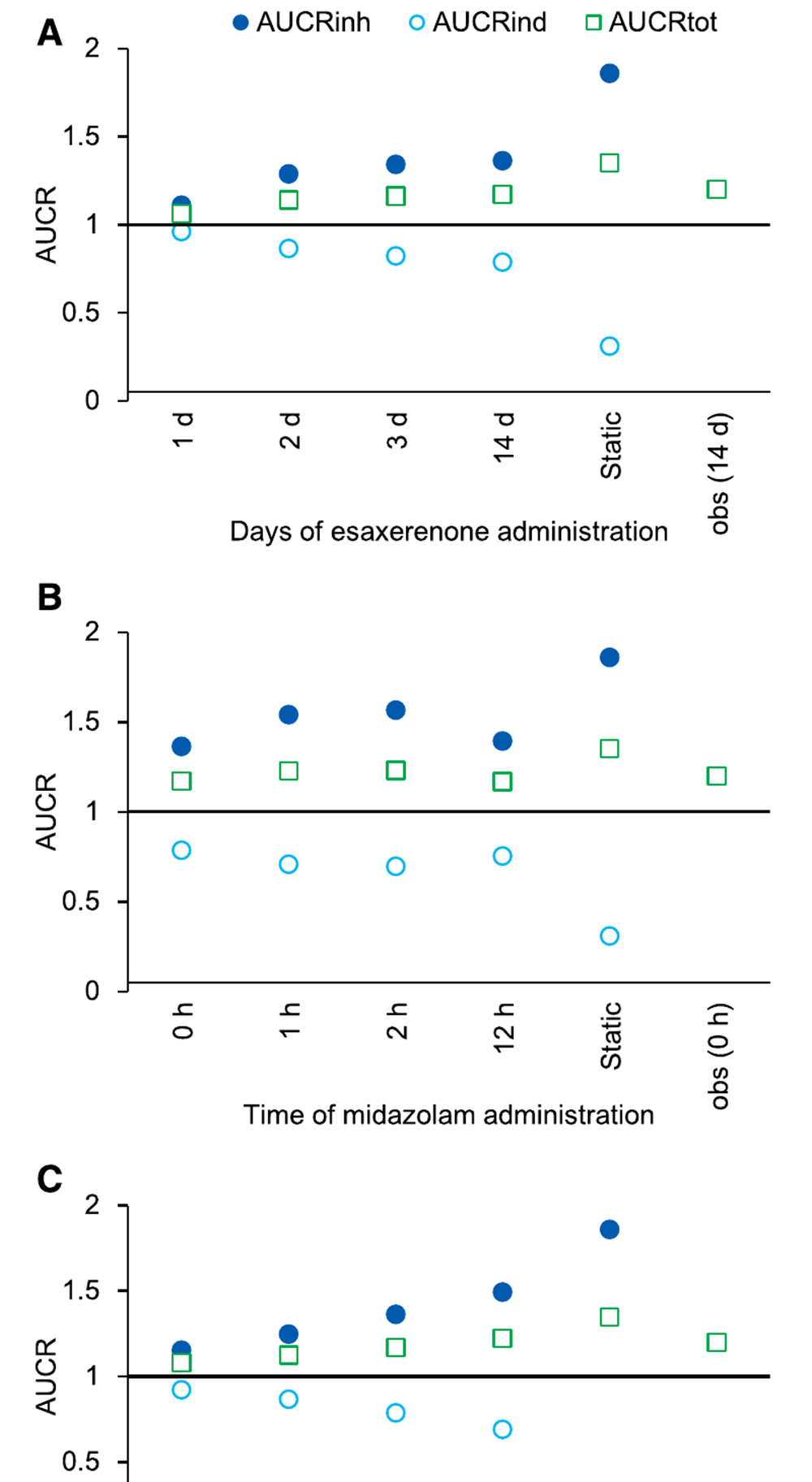
抑制与诱导的抵消效应
这篇文章最核心的一点,是作者具体展示了 抑制与诱导之间的抵消效应。当只考虑抑制时,midazolam 的 AUCR 会随着 Esaxerenone 的给药天数、剂量和给药时点而变化;当只考虑诱导时,结果则朝相反方向变化。也就是说,若分别看这两条机制,结论都可能显得比较“敏感”。
但当抑制与诱导同时纳入 PBPK 模型后,AUCRtot 始终维持在较窄范围内。无论改变给药天数、给药间隔还是剂量,总体 AUCR 变化都不大。即使将 Esaxerenone 剂量提高到 10 mg,总风险也没有明显放大。这说明 Esaxerenone 虽同时具有 TDI 和诱导作用,但两者在体内发生了部分抵消,因此其净 DDI 效应较弱。
讨论
讨论部分的重点主要有两层。第一,Esaxerenone 除 CYP3A 外,对其他主要酶和转运体的临床相关 DDI 风险总体较低。例如,文中提到其对 P-gp 的静态模型判断较低,后续与 digoxin 的临床研究也支持这一点。作者还指出,循环中的主要代谢物以 O-glucuronide 为主,而这类更高极性的代谢物通常抑制能力更弱,因此代谢物额外造成 DDI 的风险也较低。
第二,也是本文最值得注意的地方,是 静态模型与 PBPK 模型在结论上的差异。静态模型为了避免漏报风险,默认作用药物在作用部位维持较高浓度,因此更容易给出偏保守的判断。对于 Esaxerenone 这种同时存在弱 TDI 和弱诱导的药物,这种保守性会被进一步放大。相比之下,PBPK 模型能够把溶解、吸收、肠道局部浓度、酶周转及给药时间等动态因素放进同一框架,更适合回答“真实临床净效应到底有多大”这个问题。
作者还专门比较了三套不同来源的人体 PK 模型,结果显示:即使缺乏完整人体静脉数据,只要对口服吸收和肠道局部暴露的刻画合理,PBPK 仍然可以对这类弱 DDI 给出较稳定预测。这一点对 PBPK 在药物研发早期的应用很有参考意义。文中还拿 ritonavir 等同时具有抑制和诱导作用的药物作比较,说明抑制和诱导在不同给药时程下可能发生不同程度的抵消,而 Esaxerenone 的特点是两种机制都较弱,因此总 AUCR 变化较小。
另外,作者进一步讨论了不同 victim drug 场景。对于 Fg 更低、更加依赖肠道首过代谢 的 CYP3A 底物,理论上更容易受到肠道 CYP3A 变化的影响;但由于 Esaxerenone 并不会像强抑制剂那样完全抑制肠道 CYP3A,且存在诱导抵消,因此作者认为总体风险仍偏低。只是这类场景的真实临床数据目前仍较少,后续还需要更多实例来验证。
结论
Yamada 等的研究表明,Esaxerenone 在体外同时具有 CYP3A 可逆抑制、时间依赖性抑制和 CYP3A4 诱导潜力。若按传统思路分别评估这些机制,则会提示其 DDI 风险不能忽视;但在 PBPK 模型中联合纳入多种机制,并考虑肠道局部暴露后,预测的 midazolam AUCR 约为 1.2,与临床结果一致。
总体来看,Esaxerenone 虽然存在 CYP3A 相互作用信号,但其作为 perpetrator 的临床 DDI 风险较低。其核心原因在于:相互作用主要发生在肠道,且抑制与诱导在体内发生了部分抵消。从 PBPK 角度看,这篇文章也说明了:对于这类“既有抑制又有诱导”的弱 CYP3A perpetrator,PBPK 模型比传统静态模型更适合判断真实临床风险。


文章评论