摘要
这份 《药物相互作用研究技术指导原则(试行)》 不是在讨论某一个具体药物的 DDI,而是在给研发团队一套更上游的判断框架:什么时候先做体外,什么时候要做临床,什么时候可以借助基础模型、静态机制模型或 PBPK 模型来支持决策。如果把它压缩成一句话,我会说:DDI 研究不是“做几个试验交差”,而是要围绕机制、风险、阈值和临床意义,逐步把证据链搭起来。
我觉得这份文件最值得反复看的点有三个。第一,它把 DDI 研究真正拆成了一条策略链,而不是若干孤立试验。第二,它明确给出了很多实操阈值,比如代谢酶贡献 ≥25%、肾主动分泌或特定清除通路贡献 ≥25%、代谢产物总药理活性贡献 ≥50% 等。第三,PBPK 在这份文件里不是装饰性的“高级方法”,而是明确被放进从体外到临床之间的过渡工具箱里。
引言
文件开头讲得很直白:临床上患者经常联合用药,而药物-药物相互作用(Drug-drug interaction,DDI)既可能带来严重不良反应,也可能直接改变疗效。所以 DDI 的核心任务并不只是“证明会不会发生”,而是要回答更完整的问题:是否会发生、影响程度有多大、有没有临床意义、需要怎样防控,以及最后说明书该怎么写。
这也决定了这份指导原则的视角很监管。它不是从某一种技术出发,而是从 在研药物临床开发与上市信息完整性 出发。文件甚至明确提醒:DDI 研究不充分,可能妨碍获益-风险评估,也可能导致说明书受限,甚至推迟上市许可。所以这不是“可做可不做”的边角研究,而是开发策略的一部分。
研究方法
这份指导原则把 DDI 评价分成两大块:体外研究 和 临床研究。体外研究先用于识别机制,包括主要消除途径、相关代谢酶和转运体贡献、药物对酶和转运体的抑制或诱导作用;在此基础上,再决定是否需要进一步临床验证。文件特别强调,体外结果和临床药动学资料可以进入模型预测环节,这里明确列出了三类工具:基础模型、静态机制模型、动态机制模型(如 PBPK)。
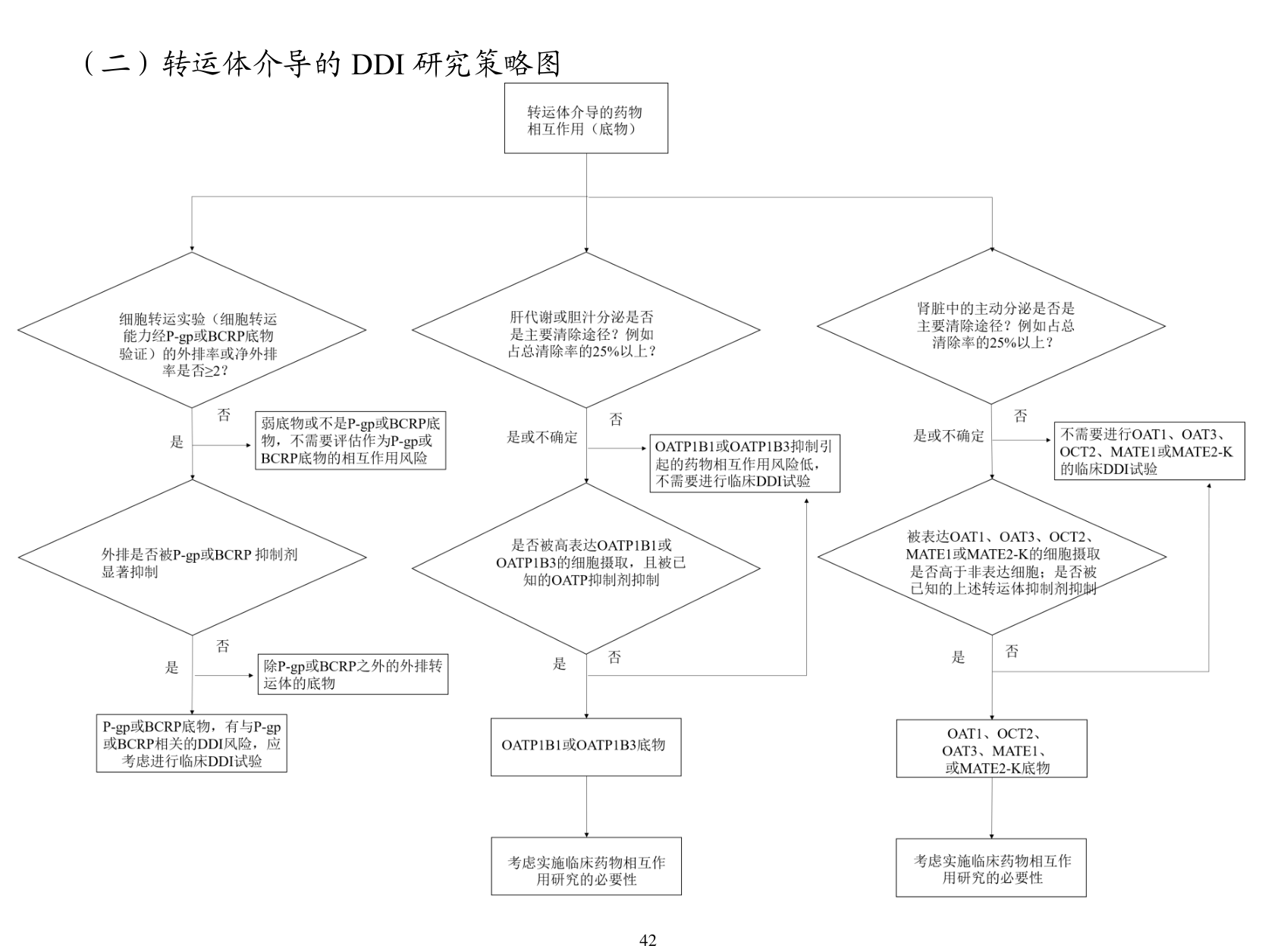
具体到代谢酶部分,文件要求优先评估主要 CYP 同工酶 CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6、CYP3A,并在必要时扩展到 FMO、MAO、AO、CES、UGT、SULT 等其他酶。对转运体部分,则重点关注 P-gp、BCRP、OATP1B1/1B3、OAT1/3、OCT2、MATE1/2-K 等常见通路。临床研究方面,文件把研究分成 前瞻性独立 DDI、前瞻性嵌套 DDI、回顾性评估,也讨论了 指针药物研究、鸡尾酒底物研究 以及特定人群设计。
如果把这整份文件当成方法学主线来看,它其实反复在强调同一件事:先用机制和阈值判断风险,再用模型和临床试验把“不确定性”缩到足够小。这也是这份指导原则最像现代 DDI 研发思维的地方。
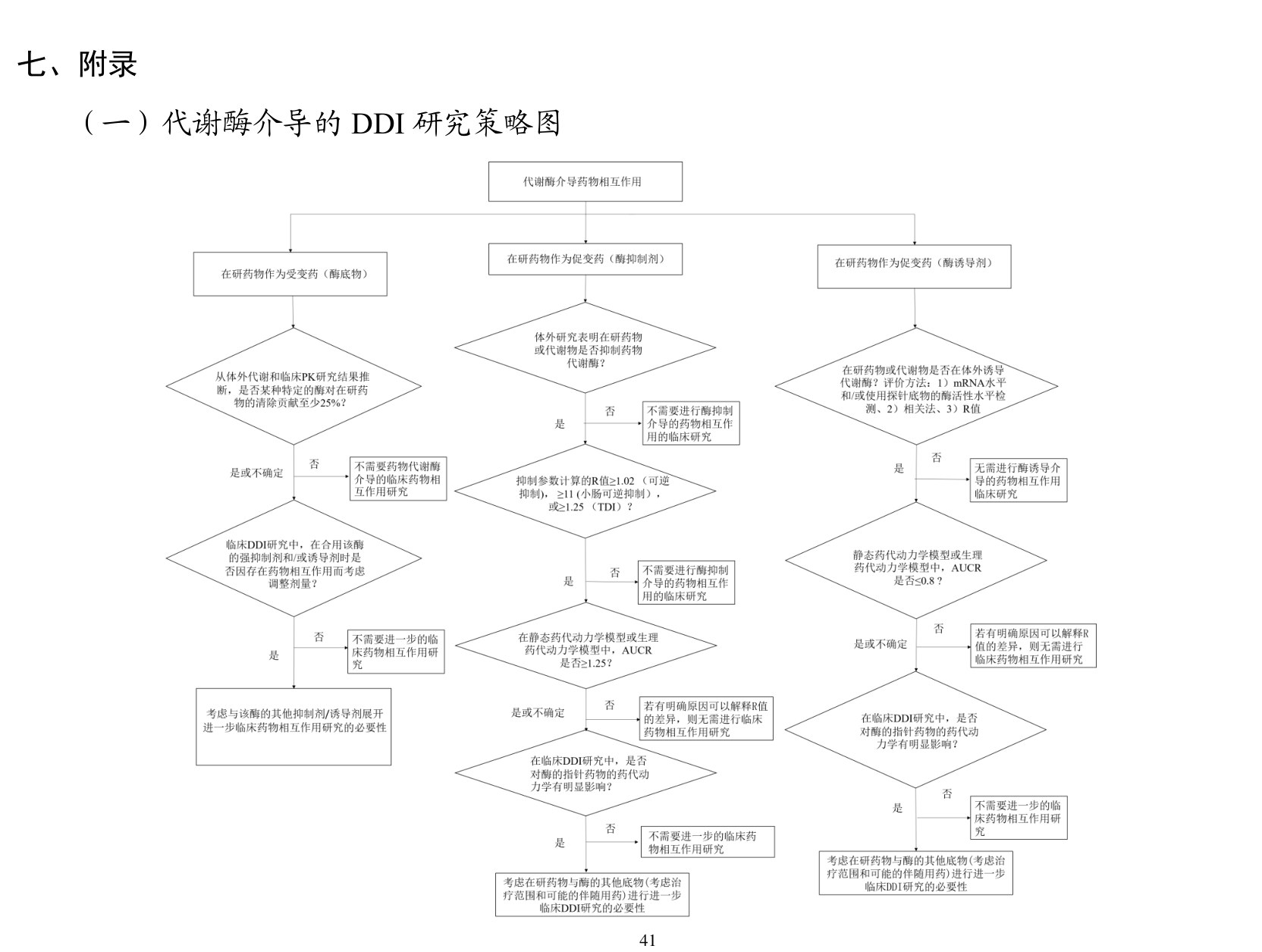
结果
如果把这份指导原则当成“监管结论”来看,我觉得最关键的结论是:DDI 研究必须围绕临床意义来分层。比如在研药物如果通过体外代谢表型研究和人体 PK 数据显示,某一特定代谢酶对总清除贡献 ≥25%,文件就认为这个酶对清除有显著贡献,此时应使用该酶的强指针抑制剂和/或诱导剂进行临床 DDI 研究。这条阈值非常重要,因为它把“要不要进临床”从模糊判断变成了比较明确的决策节点。
类似的阈值在转运体部分也出现了。比如文件提到,当肾主动分泌过程对清除较为重要,通常以 ≥25% 总消除 作为重要性的参照时,就应认真考虑 OAT1、OAT3、OCT2、MATE1/2-K 相关 DDI 风险。代谢产物部分则更细:当代谢产物对总药理活性贡献 ≥50%,或者在某些极性和暴露条件下 AUC 代谢产物达到原形药的 25% 或以上,就需要认真评估其 DDI 风险。换句话说,文件并不只关心原形药,暴露高或活性显著的代谢产物也被纳入同一套风险框架。
另外一个很有价值的结果,是文件对模型法的位置摆得很清楚。它没有把 PBPK 神化,而是给了一个很理性的层级:基础模型最简单、数据要求最少;静态机制模型更细;PBPK 数据要求最高,通常还需要临床试验数据验证,但预测能力也最强。我觉得这句话特别值得记住,因为它很像监管对 PBPK 的真实态度:PBPK 不是用来替代思考的,它是用在问题足够复杂、数据也足够支撑的时候。
PBPK 和模型预测在这份指导原则里的位置
这份文件里我最关注的一段,其实就是附录中关于模型预测的部分。文件明确写到:不同机制引起的 DDI 可以采用数学模型进行预测,常见模型包括基础模型、静态机制模型和 PBPK 模型。这说明在监管视角下,PBPK 并不是一个“额外的 bonus”,而是 DDI 评价路径中的正式工具。
但它同时也讲得很克制。基础模型可以先算 R1、R1,gut、R2、R3 这类指标,用来判断可逆抑制、小肠可逆抑制、TDI 和诱导风险;如果基础模型仍不能排除 DDI,则可以进一步走静态机制模型或 PBPK。也就是说,PBPK 不是起点,而往往是当简单方法不足以解释问题时的升级路径。这点和很多实际项目很像:不是所有 DDI 都值得上 PBPK,但一旦涉及多机制、时间依赖效应、肠肝双位点作用或需要外推到未研究场景时,PBPK 的价值就会明显上升。
文件后面关于结果外推的表述也很关键:不可能把所有临床可能合用药物都逐一做临床 DDI,因此应尽可能外推到未知情景。它举的例子很典型:如果与强效 CYP3A4 指针抑制剂合用时在研药物暴露无明显变化,通常可推断其他强、中、弱 CYP3A4 抑制剂也不太可能造成临床显著影响;但如果强抑制剂研究结果为阳性,则不一定能直接推出中弱抑制剂影响,这时就可能需要进一步临床研究或 利用 PBPK 模型评价 DDI。这几乎就是 PBPK 在监管端最典型的应用场景之一。
快速对照表
| 问题 | 指导原则给出的核心判断 | 我理解的实务含义 |
|---|---|---|
| 某代谢酶是否重要 | 对总清除贡献 ≥25% | 达到这个量级,基本就该认真设计相应指针药物 DDI 临床研究 |
| 某肾主动分泌或特定清除通路是否重要 | 常以 ≥25% 总消除 作为参考 | 转运体风险不该只停留在体外,可能需要临床确认 |
| 代谢产物是否值得单独评估 | 总药理活性贡献 ≥50%,或达到特定暴露阈值 | 不要只盯原形药,暴露高或活性强的代谢产物也可能主导 DDI 风险 |
| 什么时候用模型 | 基础模型不够时,升级到静态机制模型或 PBPK | PBPK 更像“复杂问题的升级工具”,不是所有项目的默认起点 |
| 临床 DDI 能否完全覆盖所有合并用药 | 不能,应进行合理外推 | 模型和机制理解的真正价值,在于替代不可能穷尽的全部临床组合 |
讨论
这份指导原则真正高明的地方,不在于列了多少酶和转运体,而在于它把 DDI 从“单个试验问题”变成了“整体开发策略问题”。文件反复强调的其实是:先识别风险机制,再量化风险大小,再决定需不需要临床验证,最后再考虑如何向未研究场景外推。这比“看到 CYP3A4 就机械做试验”成熟得多。
我另一个很喜欢的点,是它没有把体外、临床和模型割裂开。很多时候大家会把这三类工作当成三个部门各干各的事,但这份文件的逻辑不是这样。它更像是在说:体外研究提供机制,临床研究提供确认,模型把两者连接起来,并帮助你在研究数量有限的前提下做出更完整的判断。从这个意义上说,这份文件虽然是 DDI 指导原则,但也很像一份 PBPK 能力边界说明书。
当然,它也保留了监管应有的克制。PBPK 虽然被明确写进文件,但始终伴随着“需要足够数据支持”“通常需要临床验证”“应根据预测目的和可用数据选择模型”这些限定语。这一点我很认同,因为 模型之所以有用,前提不是模型复杂,而是问题和证据足够匹配。
结论
如果把这份指导原则压缩成一句最有用的话,我会写成:DDI 研究的核心不是把所有组合都做一遍,而是用机制、阈值、模型和临床证据,找到真正需要关注的风险并给出可执行的用药建议。
如果再换成更个人笔记一点的说法,就是:这份文件把“怎么从体外一路走到说明书”这件事讲清楚了。对做 PBPK、做临床药理、做注册策略的人来说,它不是只适合查某个阈值,而是适合反复拿来校准整个 DDI 研究思路。
我的几点启发
1. DDI 研究最怕的不是试验少,而是没有主线。 这份文件最大的价值,就是把“底物、抑制剂、诱导剂、代谢产物、模型、临床确认、结果外推”串成一条线。
2. PBPK 在 DDI 里最有价值的地方,不是替代一切临床,而是替代那些不可能穷尽的临床组合。
3. 阈值的意义不只是方便决策,更是方便团队对齐。 比如 25%、50% 这些节点,会直接决定你后面是继续做、停在体外,还是升级到模型和临床。
原文信息
药物相互作用研究技术指导原则(试行),国家药品监督管理局药品审评中心,发布时间 2021 年 1 月。这是一份 DDI 方法学与监管实践导向文件,不是单一药物的研究论文。

